营养保健食品的“蓝帽”备案注册离不开规范的临床前研究,其关键价值在于通过科学实验验证产品的功效与安全性。杭州环特生物科技股份有限公司构建了完善的临床前研究体系,为保健食品企业提供涵盖24项允许声称功能的检测服务。在临床前功效验证中,通过斑马鱼模型、哺乳动物模型等多种实验体系,量化评估产品的抗氧化、辅助降血脂、增强人体免疫能力等功效,确保功效宣称有充分的科学依据;安全性评价则聚焦急性经口毒性、遗传毒性等指标,排查产品潜在风险,保障消费者食用安全。临床前研究的数据不仅是产品备案的硬性要求,更是企业赢得市场信任的核心竞争力。环特生物凭借专业的临床前研究能力,帮助企业高效完成备案流程,推动产品快速合规上市。临床前研究数据是药物进入临床试验的关键依据。宁波候选成药分子临床前安全性
对新药临床前毒理学研究结果的准确分析与解读直接关系到新药研发的决策。在分析毒理学数据时,首先要关注各项观察指标的变化趋势,不仅只是某个时间点的数值。例如在长期毒性试验中,体重变化曲线若呈现持续下降趋势,可能意味着药物对动物的营养代谢产生了不良影响,需要进一步深入分析原因。同时,要对不同剂量组之间的数据进行对比,判断剂量 - 反应关系是否存在。若随着药物剂量的增加,毒性反应的发生率和严重程度也相应增加,那么这种剂量 - 反应关系的存在提示药物毒性与剂量密切相关。此外,对于一些异常的检测结果,不能孤立地看待,需要综合考虑动物的整体状况、其他相关指标的变化以及试验过程中的各种因素。例如,血液生化指标中某个酶活性的升高,可能是药物直接作用于肝脏细胞导致,也可能是由于动物在试验过程中受到应激等其他因素干扰。只有通过多方面、系统、科学的分析与解读,才能从毒理学研究结果中提炼出真实、准确的信息,为新药能否进入临床试验以及后续的研发方向提供合理的判断依据。杭州眼科药临床前研究服务平台环特生物临床前实验数据,可用于 NMPA 临床试验申报.
小分子药物临床前研究的关键目标是验证靶点生物学功能、明确药物作用机制,并为后续开发提供科学依据。靶点验证通常结合基因编辑技术(如CRISPR-Cas9)与细胞模型,通过敲除或过表达目标基因,观察细胞表型变化。例如,在BRAF突变型黑色素瘤研究中,研究者利用CRISPR敲除BRAF基因后,发现细胞增殖明显受抑,而回补突变型BRAF则恢复增殖,证实了该靶点的致ancer性。机制探索则依赖蛋白质组学、代谢组学等技术,解析药物对信号通路的调控。如EGFR抑制剂吉非替尼通过抑制下游AKT/mTOR通路,诱导肿瘤细胞凋亡,这一机制在临床前模型中得到验证,为后续临床试验设计提供了关键理论支持。此外,类organ模型因其保留患者tumor组织异质性的特性,成为机制研究的理想工具,可模拟药物在复杂微环境中的动态作用。
体外药效评估是临床前研究的起点,通过高灵敏度技术(如荧光标记、流式细胞术)量化候选药物对靶点的直接作用。针对激酶抑制剂,常用酶联免疫吸附试验(ELISA)或表面等离子共振(SPR)测定其对靶酶的抑制活性(如IC50、Ki值);针对抗体药物,则通过流式细胞术检测其与抗原的结合亲和力(KD值)。细胞水平实验进一步验证药物对疾病相关细胞的功能影响,例如:抗tumor药物需在多种ancer细胞系(如A549肺ancer细胞、MCF-7乳腺ancer细胞)中测试增殖抑制率(通过MTT法或Brdu掺入法);抑炎药物需在巨噬细胞中检测炎症因子(如TNF-α、IL-6)的分泌抑制效果。此外,3D细胞模型(如tumor球体、类organ)可模拟体内微环境,更真实地反映药物穿透性及细胞间相互作用。例如,某EGFR抑制剂在2D细胞实验中IC50为10nM,但在3Dtumor球体中需50nM才达同等效果,提示需优化结构以提升穿透性。环特生物深耕临床前实验,为药物研发筑牢早期研究基础.

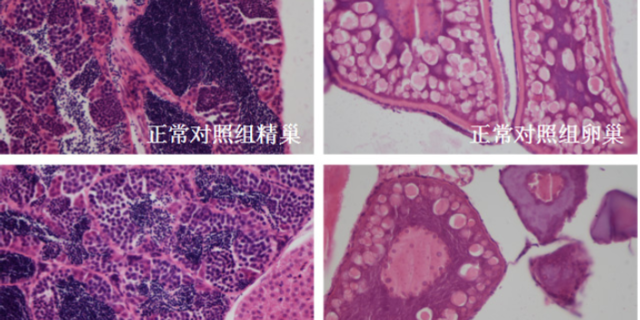
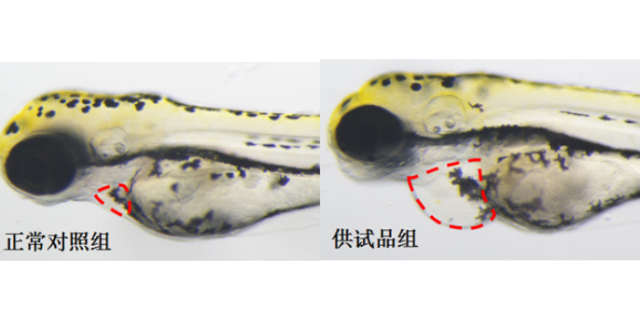
毒理学评价旨在识别药物的潜在毒性,包括急性毒性、慢性毒性、遗传毒性及生殖毒性等。传统方法依赖大鼠、犬等哺乳动物模型,但存在周期长、成本高的局限。斑马鱼模型因其胚胎透明、发育快速,成为急性毒性筛选的优先。例如,OECD指南将斑马鱼胚胎急性毒性测试(FET)纳入标准方法,通过观察胚胎死亡率、畸形率,72小时内可完成初步评估。遗传毒性评价采用Ames试验(细菌回复突变)或小鼠淋巴瘤细胞试验,检测药物是否诱发基因突变。生殖毒性研究则通过斑马鱼胚胎发育毒性测试,评估药物对心脏、神经系统的发育影响。环特生物建立的“斑马鱼-类organ”联合毒理平台,可模拟药物对肝、肾等organ的特异性损伤,提高毒性预测的准确性。例如,某候选药物在类organ中显示肝细胞凋亡率升高30%,而在斑马鱼模型中观察到肝区荧光减弱,两者结合提示需调整剂量或结构。依托关键技术,环特生物为企业定制个性化临床前实验方案。杭州眼科药临床前研究服务平台
环特生物为化妆品研发,提供专业临床前实验安全评价。宁波候选成药分子临床前安全性
候选成药分子的临床前研究是药物开发链条中的关键环节,其关键目标是通过系统评估分子的安全性、有效性及药代动力学特性,为后续临床试验提供科学依据。这一阶段需回答三个关键问题:分子是否具备医疗潜力?是否安全可控?能否在目标组织中达到有效浓度?研究内容涵盖体外活性筛选(如酶抑制、细胞增殖实验)、体内药效验证(如疾病动物模型)、毒理学评估(急性/慢性毒性、遗传毒性)及药代动力学(ADME:吸收、分布、代谢、排泄)分析。例如,针对阿尔茨海默病的候选分子Aβ寡聚体抑制剂,临床前需在转基因小鼠模型中验证其能否改善认知功能,同时通过肝微粒体孵育实验评估其代谢稳定性。这一阶段的成功标准是获得“安全有效”的初步证据,支持向IND(新药临床研究申请)申报迈进,其决策准确性直接影响药物开发成功率(据统计,临床前研究充分的分子进入临床后的成功率可提升40%)。宁波候选成药分子临床前安全性