药代动力学(PK)研究通过测定血药浓度-时间曲线,明确分子的吸收(如口服生物利用度)、分布(如血脑屏障穿透性)、代谢(如CYP450酶代谢途径)及排泄(如肾清理率)特性。例如,针对中枢的神经系统疾病候选分子,需通过脑脊液采样评估其能否穿透血脑屏障(脑/血比值>0.1为达标);针对口服药物,则需优化剂型(如纳米晶、自微乳化系统)以提升生物利用度(从<10%提升至>50%)。转化医学研究则进一步将临床前数据与临床关联——例如,通过肝微粒体孵育实验发现候选分子主要经CYP3A4代谢,提示其可能与利福平(CYP3A4诱导剂)存在药物相互作用风险,需在临床试验中设置药物相互作用研究组。此外,基于生理的药代动力学模型(PBPK)可预测不同人群(如儿童、肝肾功能不全患者)的PK特征,为临床剂量设计提供依据。这一阶段的成功标志是获得“安全、有效、可预测”的PK/PD(药效动力学)特征,为Ⅰ期临床试验的剂量爬坡设计提供关键参数。选择环特生物的临床前服务,为新药研发筑牢坚实基础。北京国家认可临床前实验室
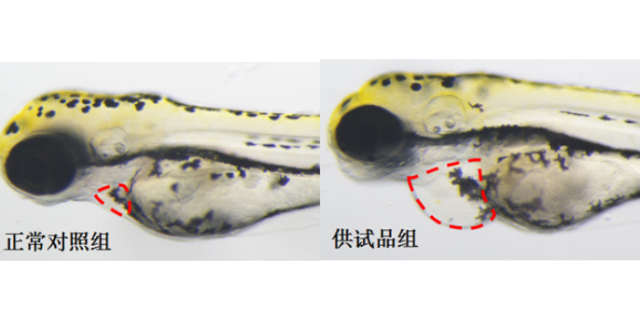
毒理学研究是临床前研究的“安全阀”,需通过急性毒性(单次高剂量给药)、重复给药毒性(28天/90天多次给药)、遗传毒性(Ames试验、小鼠淋巴瘤试验)及生殖毒性(胚胎致死性、致畸性)实验,多方面评估分子的安全性。例如,针对抗凝血药物,需通过大鼠尾静脉出血时间实验确定耐受剂量(MTD),避免引发过度出血风险;针对抗tumor药物,则需关注骨髓抑制(通过血常规检测白细胞、血小板计数)及肝毒性(通过ALT/AST酶活性测定)。特殊毒性研究(如光毒性、心脏毒性)也不可忽视——例如,通过hERG通道抑制实验评估药物是否可能引发QT间期延长。毒理学数据的解读需结合医疗窗(有效剂量与毒性剂量的比值),若候选分子的医疗指数(TI)>5,则认为安全性可控;若TI<3,则需重新优化结构或调整给药的方案。cro临床前项目报价环特生物为化妆品研发,提供专业临床前实验安全评价。
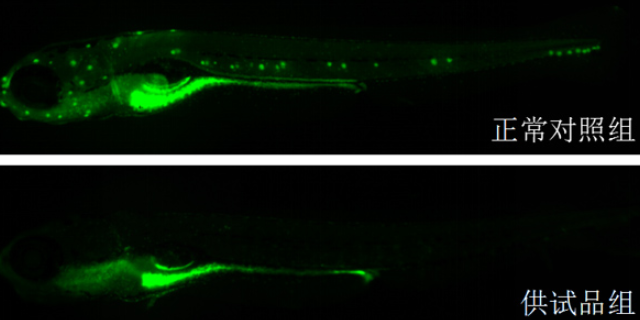
v类organ(Organoids)和器官芯片(Organ-on-a-Chip)技术是临床前药效研究的改变性工具,可模拟人体organ的复杂结构与功能。类organ由患者来源的干细胞或成体细胞在体外自组装形成,保留了原始组织的细胞类型、空间排列及部分生理功能。例如,结直肠ancer类organ可保留患者tumor的突变特征(如KRAS、APC突变),用于测试靶向药物的敏感性;肝类organ可模拟药物代谢过程,预测肝毒性。器官芯片则通过微流控技术将多种细胞类型(如内皮细胞、免疫细胞)共培养于芯片上,构建动态生理环境。例如,肺芯片可模拟呼吸运动及气流对药物分布的影响,用于评估吸入制剂的疗效。此类技术相比传统动物模型更具人源化特征,可减少种属差异导致的假阴性/阳性结果。例如,某抗纤维化药物在动物模型中无效,但在肺类organ中明显抑制成纤维细胞活化,终通过类organ数据支持其进入临床试验。
代谢性疾病(如糖尿病、肥胖、脂肪肝)的高发,推动了相关药物的研发需求,而体系化的临床前研究是药物研发成功的保障。杭州环特生物科技股份有限公司构建了覆盖多种代谢性疾病的临床前研究体系,为药物研发提供全流程支持。在临床前模型构建方面,通过高脂饲料诱导、基因编辑等方式,构建斑马鱼与哺乳动物代谢性疾病模型,模拟疾病的病理特征;在药物筛选中,利用斑马鱼模型的高通量优势,快速筛选具有降糖、降脂、jianfei等功效的候选药物;在药效验证中,通过分子生物学检测、行为学分析等,深入评估药物的医疗效果与作用机制;在安全性评价中,多方面检测药物对代谢organ(如肝脏、胰腺)的潜在影响。环特生物的体系化临床前研究服务,为代谢性疾病药物研发提供了高效、多方面的支撑。环特生物的临床前研究团队具备丰富的行业经验。

基因医疗药物作为前沿的生物药,其临床前研究面临更高的技术要求与安全标准。杭州环特生物科技股份有限公司凭借专业的技术平台,为基因医疗药物研发提供定制化的临床前研究服务。临床前研究需重点关注基因编辑工具的特异性、安全性与有效性,通过斑马鱼模型、哺乳动物模型评估基因编辑对正常细胞的影响,避免脱靶效应引发的风险;同时,需验证基因医疗药物的递送效率与靶向性,确保药物能精细到达病灶部位发挥作用。此外,临床前研究还需建立完善的生物分布与代谢检测体系,明确药物在体内的代谢路径与蓄积情况。环特生物严格遵循国际国内相关指导原则,为基因医疗药物的临床前研究提供合规、可靠的数据支持,助力该类药物的临床转化。高效的临床前研究,能大幅缩短新药从研发到上市的周期。国内临床前研发合作
临床前医药研究是连接药物研发与临床应用的桥梁。北京国家认可临床前实验室
临床前研究的起点是体外活性筛选,通过高通量技术(如96孔板、自动化液体处理系统)从化合物库中筛选出对靶点具有抑制或活动作用的“苗头化合物”。例如,针对EGFR突变型肺ancer,通过酶联免疫吸附试验(ELISA)筛选能抑制EGFR激酶活性的小分子,初始命中率可能低至0.1%。随后,通过构效关系(SAR)研究优化分子结构——通过合成系列类似物(如改变苯环取代基、调整酰胺键位置),结合表面等离子共振(SPR)技术测定结合亲和力(KD值),逐步提升活性(如将IC50从μM级优化至nM级)。这一阶段需平衡活性与理化性质(如logP、溶解度),避免“活性陷阱”(如过度追求高亲和力导致代谢不稳定)。例如,某候选HER2抑制剂通过引入氟原子降低脂溶性,成功将半衰期从2小时延长至8小时,为后续体内研究奠定基础。北京国家认可临床前实验室