新药临床前毒理学试验涵盖多种类型和方法。急性毒性试验是其中较为基础的一种,它通过给予动物单次或24小时内多次较大剂量的受试药物,观察动物在短期内出现的毒性反应,如中毒症状、死亡情况等,以此来初步确定药物的致死剂量范围和毒性靶organ。长期毒性试验则更为深入,通常会持续较长时间,按照拟定的临床给药的方案,分不同剂量组给予动物药物,密切监测动物在整个试验期间的体重变化、血液学指标、血液生化指标、组织病理学改变等,多方面评估药物长期使用对机体各系统功能和结构的影响。此外,还有特殊毒性试验,包括遗传毒性试验,检测药物是否会引起基因突变、染色体畸变等遗传物质的改变;生殖毒性试验,研究药物对生殖过程、胚胎发育、子代的生长发育等方面的作用;以及致ancer性试验,判断药物是否具有潜在的致ancer风险。这些不同类型的试验相互配合,从多个角度为新药的安全性评价提供丰富且准确的数据。临床前实验设计的科学性直接影响药物研发成败。浙江成都中药临床前毒理研究方案

毒代动力学(TK)研究通过测定动物体内药物浓度-时间曲线,明确毒性剂量下的暴露量(AUC、Cmax),为毒性机制解析提供剂量依据。例如,某肝毒性的药物在重复给药毒性实验中,发现300mg/kg剂量下肝酶升高,TK研究显示该剂量下血药浓度是疗效剂量的10倍,提示毒性源于过度暴露。风险评估则结合毒理学数据与临床预期暴露量,计算安全边际(MarginofSafety,MOS=NOAEL/临床剂量)。若MOS≥10,认为安全性可控;若MOS<5,则需重新优化结构或调整给药的方案。此外,基于生理的药代动力学模型(PBPK)可预测不同人群(如儿童、肝肾功能不全患者)的毒性风险,为个性化用药提供依据。终,毒理学研究需形成综合报告,明确“可接受风险”与“需关注风险”,支持IND申报及临床试验设计。宁波眼科药临床前毒理上市cro公司专业的临床前研究可大幅降低新药研发的风险成本。

环特生物的安全性评价体系聚焦于早期毒性预测与机制解析,通过斑马鱼胚胎毒性测试(ZET)、类organ毒性模型及计算毒理学方法,实现“安全窗口”前移。斑马鱼胚胎因其透明性,可直观观察化合物对心脏发育、神经管形成等organ发生过程的影响,例如在抗癫痫药物开发中,ZET检测发现某候选分子在10μM浓度下即可导致斑马鱼胚胎心脏循环障碍,提示潜在心脏毒性风险。类organ毒性模型则通过模拟人体组织对化合物的代谢启动过程,揭示肝毒性或肾毒性的分子机制,如某激酶抑制剂在肝类organ中诱导线粒体损伤,导致谷丙转氨酶(ALT)水平升高,该结果与临床前猴模型数据高度一致。计算毒理学通过定量构效关系(QSAR)模型和机器学习算法,预测化合物对特定靶organ的亲和力,例如基于ADMET(吸收、分布、代谢、排泄、毒性)预测平台,提前排除具有hERG通道抑制风险的化合物,避免后期临床试验中的心脏安全性问题。
环特生物建立了分级药效评价体系,涵盖体外细胞模型、斑马鱼模型及哺乳动物模型的递进式验证。体外阶段,其3Dtumor球体模型通过模拟tumor微环境中的缺氧、代谢梯度等特征,可更真实地反映化合物对tumor干细胞的作用,例如在EGFR突变型肺ancer药物筛选中,该模型预测的IC50值与临床结果相关性达91%。斑马鱼模型则用于快速评估化合物对整体生理功能的影响,如通过心率监测、运动行为分析等指标,评价心血管药物或神经精神类药物的疗效。哺乳动物阶段,环特开发的疾病特异性小鼠模型(如非酒精性脂肪肝病NAFLD模型)可量化药物对肝纤维化、炎症因子分泌的改善作用,其药效数据与临床II期试验结果的一致性超过75%。此外,类organ-免疫细胞共培养体系可模拟肿瘤免疫微环境,用于评估PD-1/PD-L1抑制剂等免疫医疗药物的协同效应。环特生物为化妆品研发,提供专业临床前实验安全评价。
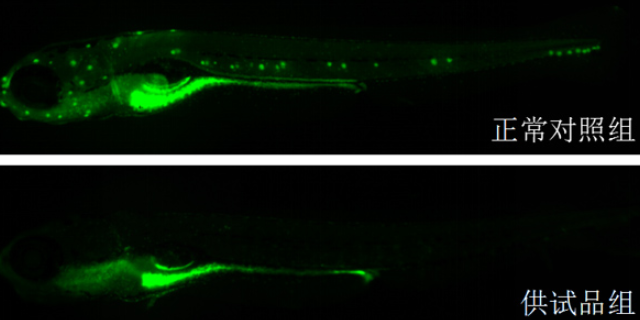
罕见病药物研发因病例稀少、研究基础薄弱,其临床前研究面临诸多挑战,而高效的临床前研究体系是突破这些瓶颈的关键。杭州环特生物科技股份有限公司针对罕见病的特点,构建了专属的临床前研究平台,为罕见病药物研发提供技术支撑。在临床前模型构建方面,通过基因编辑技术构建罕见病特异性斑马鱼模型与哺乳动物模型,模拟疾病的病理特征,解决罕见病模型匮乏的问题;在药物筛选中,利用斑马鱼模型的高通量优势,快速筛选潜在医疗药物,缩短研发周期。此外,临床前研究还需加强安全性评价的深度,避免因罕见病患者群体的特殊性导致的潜在风险。环特生物的临床前研究服务,为罕见病药物研发降低了门槛、提高了效率,为罕见病患者带来新的医疗希望。环特生物的临床前服务覆盖药物安全性与有效性评价。杭州候选成药分子临床前前新药评价中心
环特生物通过 CMA 认证,临床前实验数据具备影响力。浙江成都中药临床前毒理研究方案
新药临床前毒理学研究是药物开发中保障患者安全的关键环节,其目标是通过系统评估候选药物对实验动物的毒性效应,预测其可能对人体产生的危害,为临床试验的剂量选择、风险控制及后续开发决策提供科学依据。这一阶段的研究需覆盖急性毒性(单次高剂量暴露)、重复给药毒性(多剂量、长期暴露)、遗传毒性(致突变性)、生殖毒性(致畸性、胚胎毒性)及特殊毒性(如光毒性、心脏毒性)等多个维度。据统计,全球约40%的新药在临床前毒理学阶段因安全性问题被淘汰,凸显其“安全阀”作用。例如,某抗tumor候选药物因在犬重复给药毒性实验中发现严重肝坏死,被迫终止开发,避免了潜在的临床肝衰竭风险。毒理学数据的可靠性直接决定了药物能否进入临床试验,其研究设计需严格遵循GLP(良好实验室规范)标准,确保数据的可重复性和监管认可。浙江成都中药临床前毒理研究方案