小分子药物临床前研究的关键目标是验证靶点生物学功能、明确药物作用机制,并为后续开发提供科学依据。靶点验证通常结合基因编辑技术(如CRISPR-Cas9)与细胞模型,通过敲除或过表达目标基因,观察细胞表型变化。例如,在BRAF突变型黑色素瘤研究中,研究者利用CRISPR敲除BRAF基因后,发现细胞增殖明显受抑,而回补突变型BRAF则恢复增殖,证实了该靶点的致ancer性。机制探索则依赖蛋白质组学、代谢组学等技术,解析药物对信号通路的调控。如EGFR抑制剂吉非替尼通过抑制下游AKT/mTOR通路,诱导肿瘤细胞凋亡,这一机制在临床前模型中得到验证,为后续临床试验设计提供了关键理论支持。此外,类organ模型因其保留患者tumor组织异质性的特性,成为机制研究的理想工具,可模拟药物在复杂微环境中的动态作用。完善的质量体系,保障临床前实验全过程符合行业规范。宁波成都临床前效学评价cro
环特生物在靶点发现阶段采用多组学联合分析策略,整合基因编辑、转录组测序及蛋白质组学技术,系统挖掘疾病相关关键靶点。例如,在神经退行性疾病研究中,通过CRISPR/Cas9技术构建斑马鱼α-突触he蛋白过表达模型,结合全脑成像技术,发现特定微小RNA(miRNA)可通过调控自噬通路减缓蛋白聚集,从而锁定miR-34a作为潜在干预靶点。在验证环节,环特利用类organ模型模拟疾病病理特征,例如构建阿尔茨海默病类organ,通过单细胞测序技术揭示Aβ斑块沉积对神经元亚群的影响,为靶点功能验证提供三维组织层面的证据。此外,其开发的斑马鱼荧光报告系统可实时监测靶点活性变化,如Tg(NF-κB:EGFP)转基因斑马鱼通过绿色荧光强度量化炎症信号通路启动程度,加速了靶点验证进程。fda批准药物的临床前毒性试验临床前研究数据是药物进入临床试验的关键依据。

新药临床前毒理学研究是药物开发中保障患者安全的关键环节,其目标是通过系统评估候选药物对实验动物的毒性效应,预测其可能对人体产生的危害,为临床试验的剂量选择、风险控制及后续开发决策提供科学依据。这一阶段的研究需覆盖急性毒性(单次高剂量暴露)、重复给药毒性(多剂量、长期暴露)、遗传毒性(致突变性)、生殖毒性(致畸性、胚胎毒性)及特殊毒性(如光毒性、心脏毒性)等多个维度。据统计,全球约40%的新药在临床前毒理学阶段因安全性问题被淘汰,凸显其“安全阀”作用。例如,某抗tumor候选药物因在犬重复给药毒性实验中发现严重肝坏死,被迫终止开发,避免了潜在的临床肝衰竭风险。毒理学数据的可靠性直接决定了药物能否进入临床试验,其研究设计需严格遵循GLP(良好实验室规范)标准,确保数据的可重复性和监管认可。
候选成药分子的临床前研究是药物开发链条中的关键环节,其关键目标是通过系统评估分子的安全性、有效性及药代动力学特性,为后续临床试验提供科学依据。这一阶段需回答三个关键问题:分子是否具备医疗潜力?是否安全可控?能否在目标组织中达到有效浓度?研究内容涵盖体外活性筛选(如酶抑制、细胞增殖实验)、体内药效验证(如疾病动物模型)、毒理学评估(急性/慢性毒性、遗传毒性)及药代动力学(ADME:吸收、分布、代谢、排泄)分析。例如,针对阿尔茨海默病的候选分子Aβ寡聚体抑制剂,临床前需在转基因小鼠模型中验证其能否改善认知功能,同时通过肝微粒体孵育实验评估其代谢稳定性。这一阶段的成功标准是获得“安全有效”的初步证据,支持向IND(新药临床研究申请)申报迈进,其决策准确性直接影响药物开发成功率(据统计,临床前研究充分的分子进入临床后的成功率可提升40%)。临床前实验需严谨设计,环特生物拥有标准化实验体系.
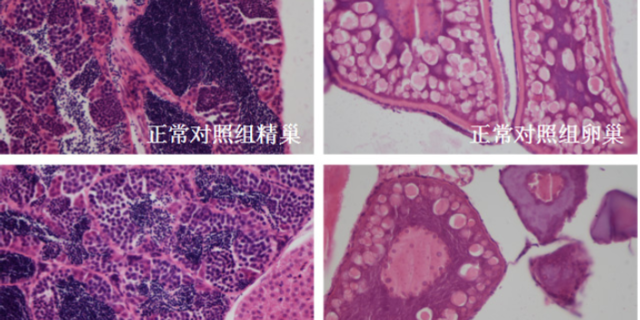
毒理学评价旨在识别药物的潜在毒性,包括急性毒性、慢性毒性、遗传毒性及生殖毒性等。传统方法依赖大鼠、犬等哺乳动物模型,但存在周期长、成本高的局限。斑马鱼模型因其胚胎透明、发育快速,成为急性毒性筛选的优先。例如,OECD指南将斑马鱼胚胎急性毒性测试(FET)纳入标准方法,通过观察胚胎死亡率、畸形率,72小时内可完成初步评估。遗传毒性评价采用Ames试验(细菌回复突变)或小鼠淋巴瘤细胞试验,检测药物是否诱发基因突变。生殖毒性研究则通过斑马鱼胚胎发育毒性测试,评估药物对心脏、神经系统的发育影响。环特生物建立的“斑马鱼-类organ”联合毒理平台,可模拟药物对肝、肾等organ的特异性损伤,提高毒性预测的准确性。例如,某候选药物在类organ中显示肝细胞凋亡率升高30%,而在斑马鱼模型中观察到肝区荧光减弱,两者结合提示需调整剂量或结构。环特生物开展基因编辑研究,赋能临床前实验创新升级、。天然药物临床前cro企业
环特生物五大实验中心,就近承接各地临床前实验需求。宁波成都临床前效学评价cro
毒代动力学(TK)研究通过测定动物体内药物浓度-时间曲线,明确毒性剂量下的暴露量(AUC、Cmax),为毒性机制解析提供剂量依据。例如,某肝毒性的药物在重复给药毒性实验中,发现300mg/kg剂量下肝酶升高,TK研究显示该剂量下血药浓度是疗效剂量的10倍,提示毒性源于过度暴露。风险评估则结合毒理学数据与临床预期暴露量,计算安全边际(MarginofSafety,MOS=NOAEL/临床剂量)。若MOS≥10,认为安全性可控;若MOS<5,则需重新优化结构或调整给药的方案。此外,基于生理的药代动力学模型(PBPK)可预测不同人群(如儿童、肝肾功能不全患者)的毒性风险,为个性化用药提供依据。终,毒理学研究需形成综合报告,明确“可接受风险”与“需关注风险”,支持IND申报及临床试验设计。宁波成都临床前效学评价cro