根据2012年一篇文献的统计,在临床试验阶段因安全性原因失败的药物占了很大的比重。这就更加要求我们在临床前阶段要非常***谨慎地进行药物安全性评价,毕竟“KillEarly”可以尽量避免临床失败的巨大损失。急性毒性是指药物在单次或24小时内多次给予后一定时间内所产生的毒性反应。试验应采用至少两种哺乳动物进行试验,一般应选用一种啮齿类动物和一种非啮齿类动物。原则上,给药剂量应包括从未见毒性反应的剂量到出现严重毒性反应的剂量,或达到比较大给药量。推荐靠谱的药物安全性评价机构。山东专业药物安全性评价实验
长期以来,我国的生物制药申报以“生物类似药”为主,药物评价关注于候选药与原研药之间质量比对研究的“相似性”,药效毒理比较研究的“桥接性”、临床试验的“非劣性”和临床价值的“可接受性”。因此,抗体生物类似药在申请IND前应基本确定生产工艺,并进行充分的结构确证与质量研究。而创新型抗体药物研发进程具有“创试性”(临床试验存在较大的失败风险)、“阶段性”(药学研究可分阶段展开)和“渐进性”(临床期间通常发生工艺变更)的特征,其评价关注点应区别于传统生物类似药,药学研究内容可按照临床试验进展分阶段进行要求,重点关注于影响临床药物安全性评价问题。湖南高质量药物安全性评价检测非临床药物安全性评价研究是药物研发的基础性工作。

新药申请是申办者采取的要求FDA考虑批准新药在美国上市的正式步骤。在完成了Ⅲ期临床试验并获得了药物安全性评价信息后,申办者提出新药申请。现在,可以以电子方式提交临床研究报告,CDISO正是基于这一要求而开发的用于数据提交/交换的标准。NDA的审评由药理、化学、统计、医学和药物代谢等不同专业的人员组成的审核小组进行,并由消费者安全官统筹:(1)消费者安全官检查所送资料是否完整。(2)审核小组审核。(3)召集**咨询委员会会议,公开讨论试验的结果并提出委员会意见当NDA审核结束后,FDA将给申办者一份核准信函并说明所需补充的资料或手续当所有手续齐全后,经FDA同意,寄发新药上市许可核准书,作为药物上市的法律依据。
药物安全性评价,生产原材料方面,裸抗分子可参照同类品种(全新序列抗体或生物类似药)进行评价。小分子***、连接子作为ADC药物的关键原材料,应提供详细的起始物料、合成工艺、结构确证、质量控制等信息,应重点关注影响产品安全性的小分子化药杂质(可偶联、不可偶联)和有机溶剂残留。建议申请人对于含量较高的杂质(>0.1%)应进行结构鉴定或药物安全性评价,有机溶剂残留应符合按照ICHQ3A或2015版《中国药典》相关规定。生产工艺方面,应结合产品质量属性(平均载药量、聚体等)对化学偶联相关工序进行优化,通过设置工艺参数控制范围和建立中间体验收标准,保证工艺稳健性和产品质量一致性。偶联后纯化工艺一般采用凝胶过滤、切向流过滤或超滤技术等去除小分子杂质和有机溶剂。应结合工艺相关杂质的去除效果或残留量,对纯化工艺关键参数(过滤体积等)验证。药效学的研究有助于药物安全性评价。

药物安全性评价,生物药的原研制剂很多质量参数都是在一个可接受范围内,只要在该范围内的波动就不会改变药品的安全性和有效性——这也是QbD理念的**所在——不会对品质造成影响。曾有一家瑞士仿制药公司,为了获得某抗体原研药的某些关键参数的可接受波动范围,花费了巨额资金购买罗氏公司生产的、甚至至失效期的多批次样品,这些样品横跨了2008年11月到2011年4月期间的20多个不同批次的样品进行分析,**终发现有些参数在较宽范围内波动,但没有影响到药物安全性评价,从而为自我仿制制剂的开发确定了合理科学的“度”,**终使得研发工作一针见血、事半功倍!药物安全性评价要求药物的基本要求、安全、有效、质量可控 。湖南高质量药物安全性评价检测
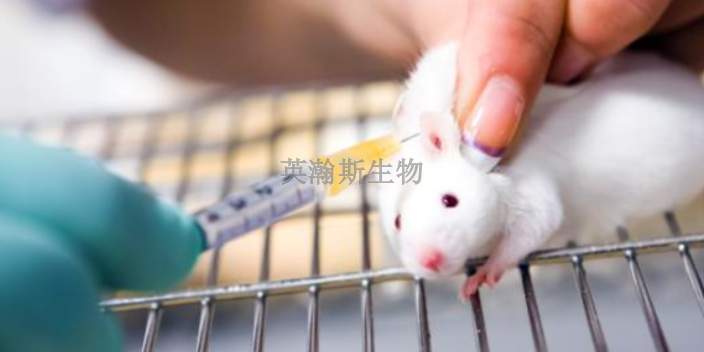
英瀚斯生物具有丰富的药物安全性评价研究经验和GLP管理经验。山东专业药物安全性评价实验
美国现有的生物技术产品监管体系能够有效保护健康和环境,在某些情况下,机构管辖范围的不确定性会带来不必要的成本和负担,如审查缺乏时间可预测性等情况。这些成本和负担限制了中小型企业应对监管程序的能力,使公众很难理解如何确保这些产品进行了药物安全性评价,进而有可能延缓经济增长,阻碍创新和竞争力。美国认为,需要进一步更新协调框架,促进**通过监管体系进行合理监管,并提高透明度,同时继续为推动创新提供科学的监管框架。山东专业药物安全性评价实验
南京英瀚斯生物科技有限公司主要经营范围是医药健康,拥有一支专业技术团队和良好的市场口碑。公司自成立以来,以质量为发展,让匠心弥散在每个细节,公司旗下实验外包,动物模型构建,细胞分子实验,病理检测深受客户的喜爱。公司将不断增强企业重点竞争力,努力学习行业知识,遵守行业规范,植根于医药健康行业的发展。英瀚斯生物凭借创新的产品、专业的服务、众多的成功案例积累起来的声誉和口碑,让企业发展再上新高。